
When I (Ben) was in grad school, I worked on a Nextflow pipeline called GEMmaker, an RNA-seq analysis pipeline similar to nf-core/rnaseq. We quickly ran into a problem, which is that on large runs, we were running out of storage! As it turns out, it wasn’t the final outputs, but the intermediate outputs (the BAM files, etc) that were taking up so much space, and we figured that if we could just delete those intermediate files sooner, we might be able to make it through a pipeline run without running out of storage. We were far from alone.
Automatic cleanup is currently the oldest open issue on the Nextflow repository. For many users, the ability to quickly delete intermediate files makes the difference between a run being possible or impossible. Stephen Ficklin, the creator of GEMmaker, came up with a clever way to delete intermediate files and even “trick” Nextflow into skipping deleted tasks on a resumed run, which you can read about in the GitHub issue. It involved wiring the intermediate output channels to a “cleanup” process, along with a “done” signal from the relevant downstream processes to ensure that the intermediates were deleted at the right time.
This hack worked, but it required a lot of manual effort to wire up the cleanup process correctly, and it left me wondering whether it could be done automatically. Nextflow should be able to analyze the DAG, figure out when an output file can be deleted, and then delete it! During my time on the Nextflow team, I have implemented this exact idea in a pull request, but there are still a few challenges to resolve, such as resuming from deleted runs (which is not as impossible as it sounds).
Many users have told me that they would gladly take the cleanup without the resume, so I found a way to provide the cleanup functionality in a plugin, which I call nf-boost. This plugin is not just about automatic cleanup – it contains a variety of experimental features, like new operators and functions, that anyone can try today with a few extra lines of config, which is much less tedious than building Nextflow from a pull request. Not every new feature can be implemented via plugin, but for those features that can, it’s nice for the community to be able to try it out before we make it official.
The nf-boost plugin requires Nextflow v23.10.0 or later. You can enable the experimental cleanup by adding the following lines to your config file:
plugins {
id 'nf-boost'
}
boost {
cleanup = true
}
The strategy of automatic cleanup is simple:
In practice, the conditions for 2(a) are tricky to get right because Nextflow doesn’t know the full task graph from the start (thanks to the flexibility of Nextflow’s dataflow operators). But you don’t have to worry about any of that because we already figured out how to make it work! All you have to do is flip a switch (boost.cleanup = true) and enjoy the ride.
Let’s consider a variant calling pipeline following standard best practices. Sequencing reads are mapped onto the genome, producing a BAM file which will be marked for duplicates, filtered, recalibrated using GATK, etc. This means that, for a given sample, at least four copies of the BAM file will be stored in the work directory. In other words, for an initial paired-end whole-exome sequencing (WES) sample of 12 GB, the work directory will quickly grow to 50 GB just to store the BAM files for one sample, or 100 GB for a paired sample (e.g. germline and tumor).
Now suppose that we want to analyze a cohort of 100 patients – that’s ~10 TB of intermediate data, which is a real problem. For some users, it means processing only a few samples at a time, even though they might have the compute capacity to do much more. For others, it means not being able to process even one sample, because the accumulated intermediate data is simply too large. With automatic cleanup, Nextflow should be able to delete the previous BAM as soon as the next BAM is produced, for each sample independently.
We tested this use-case with a paired WES sample (total input size of 26.8 GB), by tracking the work directory size for a run with and a run without automatic cleanup. The results are shown below.
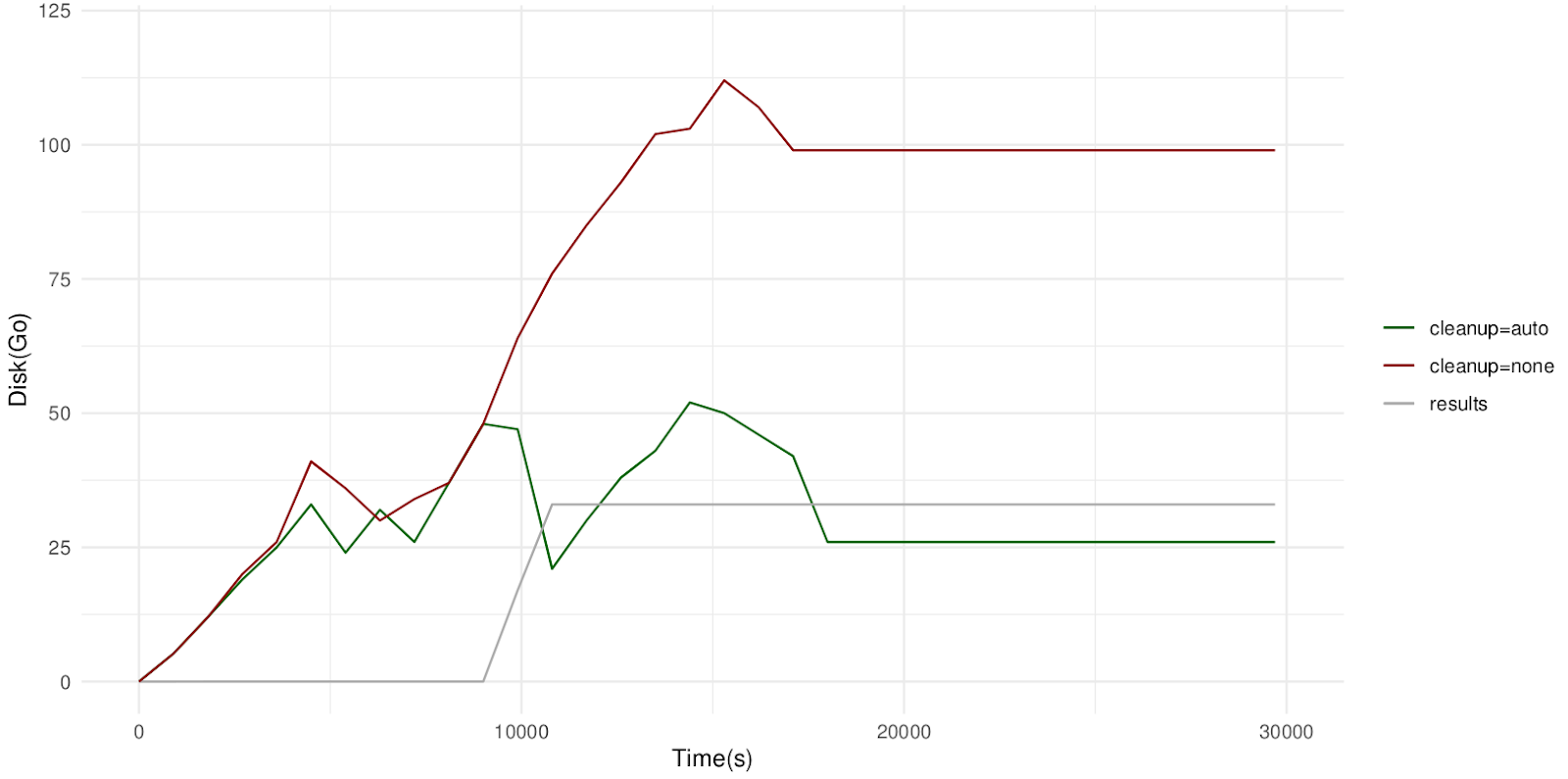
Note: we also changed the boost.cleanupInterval config option to 180 seconds, which was more optimal for our system.
As expected, we see that without automatic cleanup, the size of the work directory reaches 110 GB when all BAM files are produced and never deleted. On the other hand, when the nf-boost cleanup is enabled, the work directory occasionally peaks at ~50 GB (i.e. no more than two BAM files are stored at the same time), but always returns to ~25 GB, since the previous BAM is deleted immediately after the next BAM is ready. There is no impact on the size of the results (since they are identical) or the total runtime (since cleanup happens in parallel with the workflow itself).
In this case, automatic cleanup reduced the total storage by 50-75% (depending on how you measure the storage). In general, the effectiveness of automatic cleanup will depend greatly on how you write your pipeline. Here are a few rules of thumb that we’ve come up with so far:
Automatic cleanup in nf-boost is an experimental feature, and notably does not support resumability, meaning that the deleted files will simply be re-executed on a resumed run. While we work through these last few challenges, the nf-boost plugin is a nice option for users who want to benefit from what we’ve built so far and don’t need the resumability.
The nice thing about nf-boost’s automatic cleanup is that it is just a preview of what will eventually be the “official” cleanup feature in Nextflow (when it is merged), so by using nf-boost, you are helping the future of Nextflow directly! We hope that this experimental version will help users run workloads that were previously difficult or even impossible, and we look forward to when we can bring this feature home to Nextflow.
This post was contributed by a Nextflow Ambassador. Ambassadors are passionate individuals who support the Nextflow community. Interested in becoming an ambassador? Read more about it here.